Sequence-based stability changes prediction
SCpre-seq, a residue level 3D structure-based prediction tool to assess single point mutation effects on protein thermodynamic stability and applying to dingle-domain monomeric proteins. Given protein sequence with single mutations as the input, the proposed model integrated both sequence level features of mutant residues and residue level mutation-based 3D structure features.
SCpre-seq’s applications
SCpre-seq can be applied to predicting stability changes on single monomeric proteins which tertiary structures are unavailable.
SCpre-seq can be used to assess both somatic and germline substitution mutations such as p53 in biological and medical research on genomics and proteomics.
Mutation-based background knowledge of variants obtained from G2S which surveys the whole mutations in PDB can be used to makes up for the shortage of structural information for sequence-based methods.
A reliable mutation-based residue-level 3D structure information has applied to improve the model’s performance.
Reference
ACKNOWLEDGEMENTS
Support
Feel free to submit an issue or send us an email. Your help to improve SCpre-seq is highly appreciated.
About
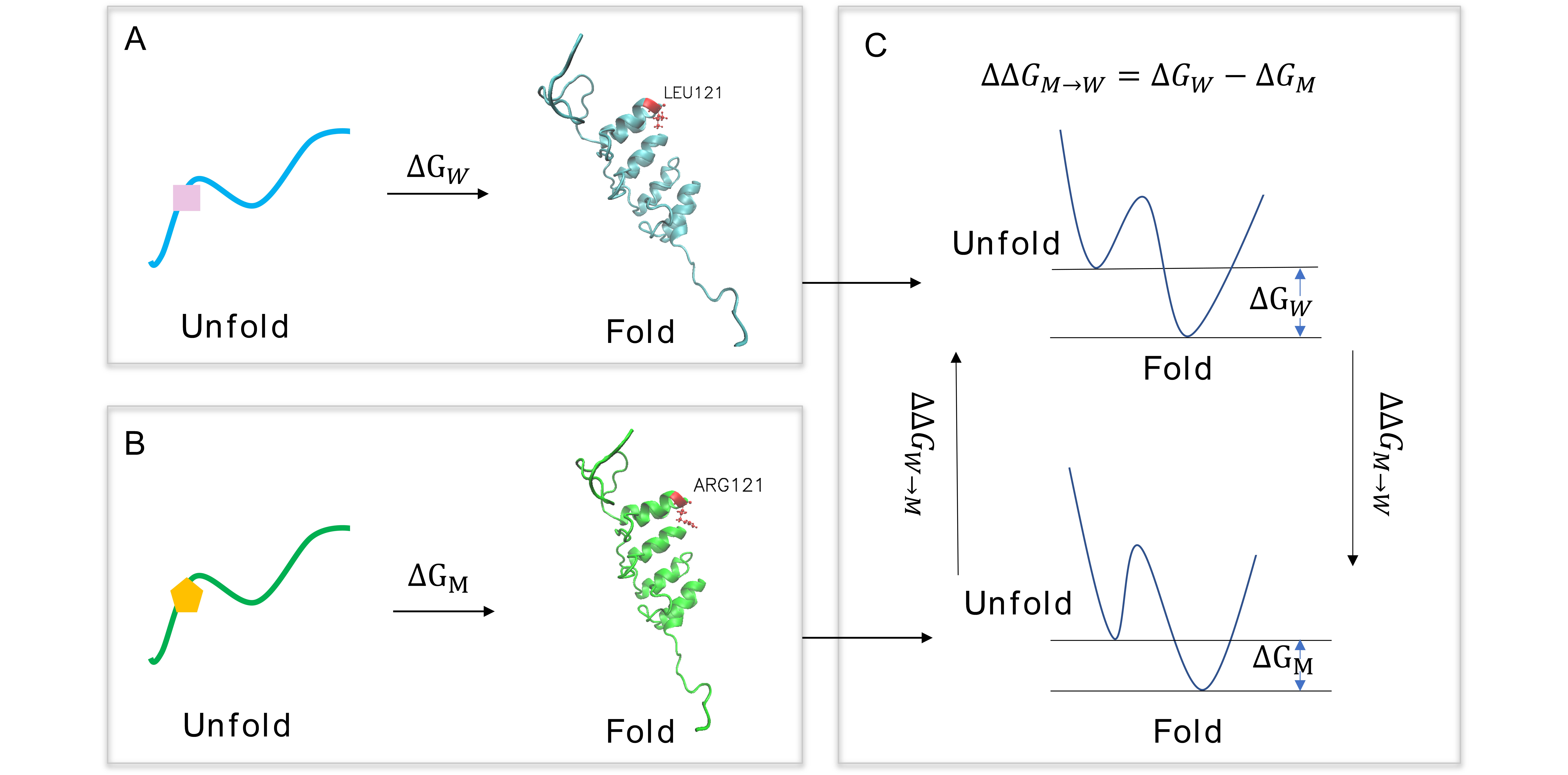
Mutation-induced protein thermodynamic stability changes (DDG) are crucial for understand protein biophysics, genomic variant interpretation, and mutation-related diseases. We introduce SCpre-seq, a residue level 3D structure-based prediction tool to assess the effects of single point mutation on protein thermodynamic stability and applying to dingle-domain monomeric proteins. Given protein sequence with single mutations as the input, the proposed model integrated both sequence-level features of mutant residues and mutation-based structure features. Our stability predictor outperformed previously published methods on various benchmarks. SCpre-seq will be a dedicated resource for assessing both somatic and germline substitution mutations in biological and medical research on genomics and proteomics.
What is DDG?
Protein thermodynamic stability changes of single point mutation are changes of the Gibbes free energy for the biophysical process of protein folding between two states before and after single point mutation on the protein[1]. A quantified change of Gibbs free energy of a protein between the folding and unfolding status is usually represented as DG. When a point mutation is present and a residue is substituted in a protein, the original protein would be a “reference state”, likely called “wild-type protein”. The protein mutated is called “mutation protein”.
Installation
Clone the package
git clone https://github.com/hurraygong/SCpre-seq.git
cd SCpre-seq
Install dependencies
1. Install Anaconda3
Download Anaconda and install. If your computer has alread installed, pass this step.
2. Install HHsuite
Using conda install hhsuite. It is also can be installed by other methods shown on official website, If you have installed, pass this step.
conda install -c conda-forge -c bioconda hhsuite
Install HHsuite database.
mkdir HHsuitDB
cd HHsuitDB
wget http://wwwuser.gwdg.de/~compbiol/uniclust/2020_06/UniRef30_2020_06_hhsuite.tar.gz
mkdir -p UniRef30_2020_06
tar xfz UniRef30_2020_06_hhsuite.tar.gz -C ./UniRef30_2020_06
rm UniRef30_2020_06_hhsuite.tar.gz
3. Install DSSP
Using conda install DSSP programe. (https://anaconda.org/salilab/dssp)
conda install -c salilab dssp
Then to find the execute programe mkdssp. You can change the mkdssp
to dssp using cp command by yourself.
whereis mkdssp
4.Install BLAST++
We also recommend to install BLAST++ using conda. To install this package with conda run one of the following:
conda install -c bioconda blast
conda install -c bioconda/label/cf201901 blast
Install BLAST++ database.
mkdir PsiblastDB/
cd PsiblastDB
wget https://ftp.ncbi.nlm.nih.gov/blast/db/swissprot.tar.gz
wget https://ftp.ncbi.nlm.nih.gov/blast/db/swissprot.tar.gz.md5
tar -zxvf swissprot.tar.gz
rm swissprot.tar.gz
rm swissprot.tar.gz.md5
Then you can proceed to the next step.
Datasets
Datasets for training, testing and casestudy
Our work used datasets S1676, S236, S543 to investigate the prediction of stability changes on protein (Table S1).
Table S1 Datasets used to build, evaluate and independently test in SCpre-seq
Dataset |
Total Variants (Proteins) |
Destabilizing Variants (Proteins) |
Stabilizing Variants (Proteins) |
Stabilizing Variants (Proteins) |
Additional Details |
|---|---|---|---|---|---|
S1676 |
1676 (67) |
1,223 (64) |
424 (53) |
29(4) |
Unique Variants/Averaged DDG |
S236 |
236 (22) |
192 (18) |
42 (14) |
2(2) |
Unique Variants/Averaged DDG |
S543 |
543(55) |
426(48) |
107(37) |
10(6) |
Unique Variants/Averaged DDG |
p53 |
42 (1) |
31(1) |
11 (1) |
0 |
One Protein |
Databases and references
Folkman, L., et al., EASE-MM: Sequence-Based Prediction of Mutation-Induced Stability Changes with Feature-Based Multiple Models. J Mol Biol, 2016. 428(6): p. 1394-1405.
Pires, D.E., D.B. Ascher, and T.L. Blundell, mCSM: predicting the effects of mutations in proteins using graph-based signatures. Bioinformatics, 2014. 30(3): p. 335-42.
Datasets with features
Downloading
Quickstart
Quikly Run scGNN
This is the parameters to run SCpre-seq.
parser.add_argument('-l', '--variant-site', dest='variant_list', type=str,required=True, help='A list of variants, one per line in the format "POS WT MUT", a file')
parser.add_argument('-w', '--variant-wildtype', dest='variant_wildtype', type=str,
required=True, help='wild-type residue, for example residue "A"')
parser.add_argument('-m', '--variant-mutation', dest='variant_mutation', type=str,
required=True, help='mutation residue, for example residue "A"')
parser.add_argument('-p', '--variant-position', dest='variant_position', type=int,
required=True, help='variant position in sequence')
parser.add_argument('-s', '--sequence', type=str,
required=True, help='A protein primary sequence in a file in the format fasta.')
# background mutation Features from G2s
parser.add_argument('--pdbpath', type=str,default='/storage/htc/joshilab/jghhd/SC/stability_change1/datasets_s1676_seq/PDB/',
required=True, help='A path for storage matched PDB structures')
parser.add_argument('--dssppath', type=str,default='/storage/htc/joshilab/jghhd/SC/stability_change1/datasets_s1676_seq/dssp/',
required=True, help='A path for storage dssp output files')
parser.add_argument('--dsspbin', type=str,default='mkdssp',
required=True, help='Execute bin path for DSSP')
parser.add_argument('--psiblastbin', type=str,default='psiblast',
required=True, help='Execute bin path for psiblast')
parser.add_argument('--hhblitsbin', type=str,default='hhblits',
required=True, help='Execute bin path for hhblits')
parser.add_argument('--psiblastout', type=str,default='./Sequence/psiout',
required=True, help='A path for storage psiblast out files')
parser.add_argument('--psiblastpssm', type=str,default='./Sequence/pssmout',
required=True, help='A path for storage psiblast pssm files')
parser.add_argument('--psiblastdb', type=str,default='/home/gongjianting/tools/PsiblastDB/swissprot',
required=True, help='background database for align in psiblast')
parser.add_argument('--hhblitsdb', type=str,default='/home/gongjianting/tools/HHsuitDB/UniRef30_2020_06',
required=True, help='background database for align in tools hhblits')
parser.add_argument('--hhblitsout', type=str,default='./Sequence/hhblitout',
required=True, help='A path for storage hhblits hhr files')
parser.add_argument('--hhblitshhm', type=str,default='./Sequence/hhmout',
required=True, help='A path for storage hhblits hhm files')
parser.add_argument("-v", "--version", action="version")
parser.add_argument("-o", "--outfile",type=bool, default=False,help='Whether save the result or not')
parser.add_argument("-printout", type=bool, default=True, help='Whether print the result or not')
parser.add_argument("-outpath", "--outfilepath", type=str,default='./',help='Output file path')
Take mutation Q to H at position 104 of p53 protein as example. Its position in sequence is 9. So the run command as follows:
python StatGetPDBlist.py -p 9 -w Q -m H -o True -s ./examples/SEQ/2ocj_A129D.fasta --pdbpath ./examples/PDBtest --dssppath ./examples/DSSPtest --dsspbin mkdssp --psiblastbin psiblast --hhblitsbin hhblits --psiblastout ./examples/psiout --psiblastpssm ./examples/pssmout --psiblastdb /home/gongjianting/tools/PsiblastDB/swissprot --hhblitsdb /home/gongjianting/tools/HHsuitDB/UniRef30_2020_06 --hhblitsout ./examples/hhblitout --hhblitshhm ./examples/hhmout -outpath ./examples/
Mutation structure preparation
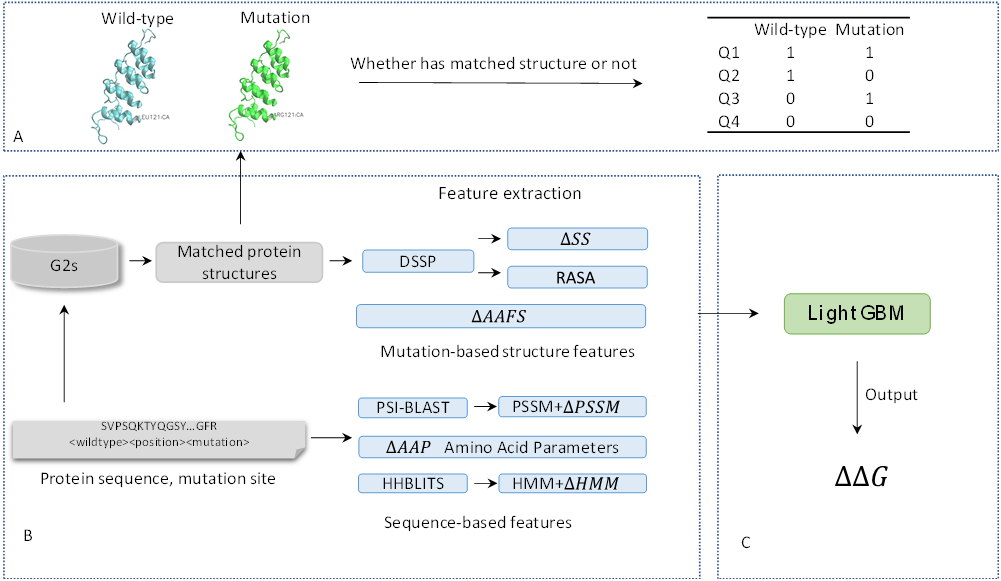
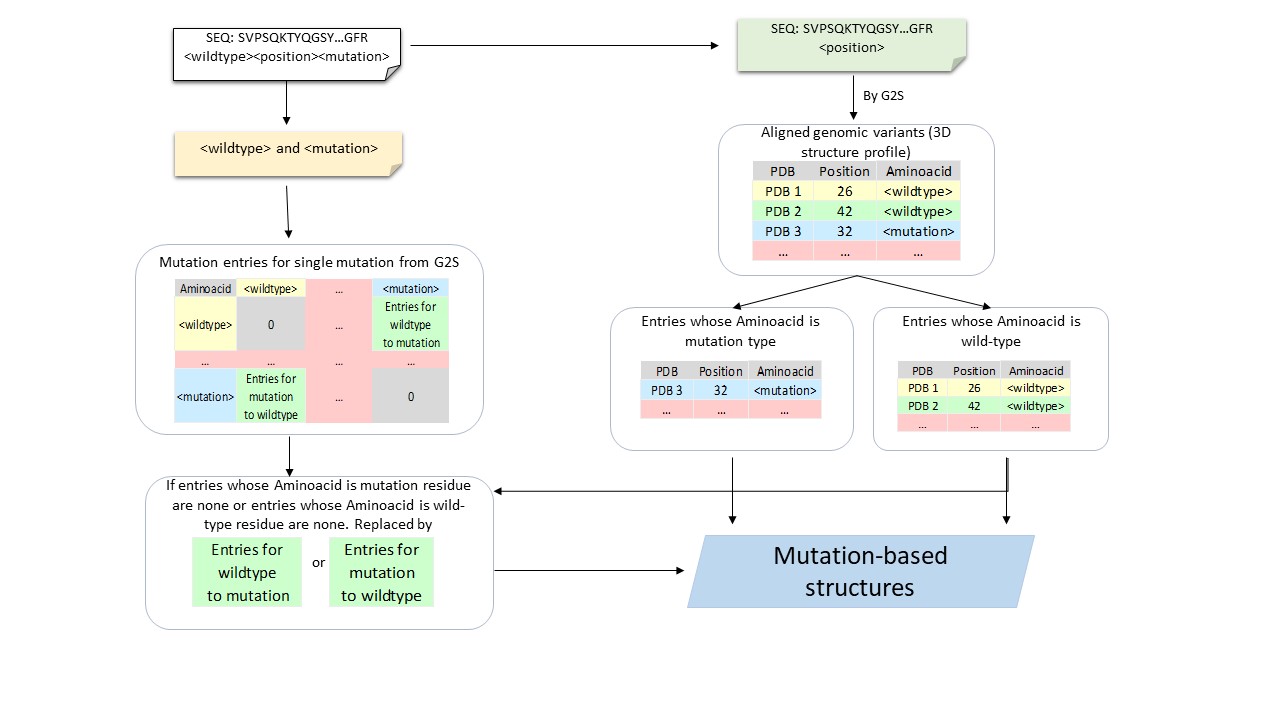
[G2S](https://g2s.genomenexus.org) provides a real-time web API that provides an automated mapping of genomic variants on 3D protein structures. Giving protein sequence and the position of a variant as the query, G2S searches to get a 3D structure profile that is a list of protein structures with active residues whose positions are matched with the position of input variant after aligning with queried sequence. We divided this list of protein structures of variants into two classes based on the type of the wild-type and mutant amino acids from the input data (Supplemental Figure S1). Due to the availabilities of the PDB structures, four categories of PDB structures are adopted. Q1: Both tertiary structures in the 3D structure profile whose active residues are matched to wild-type residue and tertiary structures in the 3D structure profile whose active residues are matched to mutant residue are available. Q2: Tertiary structures in the 3D structure profile whose active residues are wild-type residue are available while structures which matched mutant residue are unavailable. Q3: Tertiary structures in the 3D structure profile whose active residues are wild-type residue are unavailable while tertiary structures which matched mutant residue are available. Q4: Neither tertiary structure in 3D structure profile which matched wild-type residue nor tertiary structures which matched mutant residue are available.
The samples count for 4 situations when mutation-based structures are available or not on datasets S1676, S543 and S236.
Dataset |
Q1 |
Q2 |
Q3 |
Q4 |
|---|---|---|---|---|
S543 |
172 |
366 |
1 |
4 |
S236 |
46 |
159 |
0 |
31 |
S1676 |
706 |
950 |
0 |
20 |
Results Analysis
Performance on multiple models according to mutation-based structure information
To build a robust predicting protein stability changes model, dataset S1676 is used to train a model using 10-fold cross-validation. The results of our methods are replicated 10-fold cross-validation 20 times with shuffling the training data. Based on the availability of the PDB structure, we proposed four models SCpre-seq^str, SCpre-seq^seq, SCpre-seq and SCpre-seq^* to conduct comparative tests. When assuming that structures of wild-type and mutation proteins are unavailable for the training dataset, mutation-based structure features would get from G2S (SCpre-seq*).
Table 1. Performance of 10-Fold cross-validation on Dataset S1676
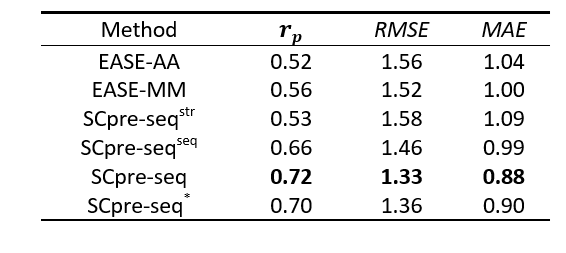

Figure 1. The Pearson correlation coefficient of 10-fold cross-validation for six models EASE-AA, EASE-MM, SCpre-seq^str, SCpre-seq^seq, SCpre-seq and SCpre-seq^* on the training dataset
SCpre-seq achieves state-of-the-art performance on testing sets
To evaluate the robustness of our model, we used S236, S543 as the independent testing datasets. SCpre-seq compared with nine commonly used methods including EASE-AA, EASE-MM, MUpro, I-Mutant2.0, INPS, SAAFEC-SEQ, DDGun and PoPMuSiC, MAESTRO on different testing datasets. Among them, EASE-AA, EASE-MM(EASE-MM-web), MUpro, sequence-based version of I-Mutant2.0(I-MutantA), INPS, SAAFEC-SEQ, DDGun predict ∆∆G starting from sequence and mutation residues while the structure-based version of I-Mutant2.0(I-MutantB), PoPMuSiC, MAESTRO required structures as input. The results are shown in Figures 2 and 3 for S236 and S543, respectively.
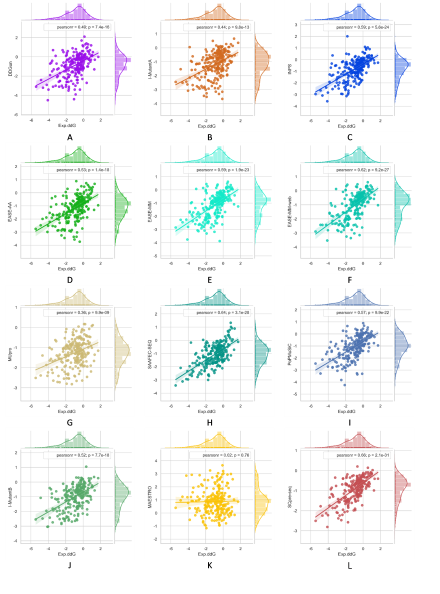
Figure 2. Multiple bivariate plots for 11 comparative methods and SCpre-seq with marginal histograms on dataset S236.
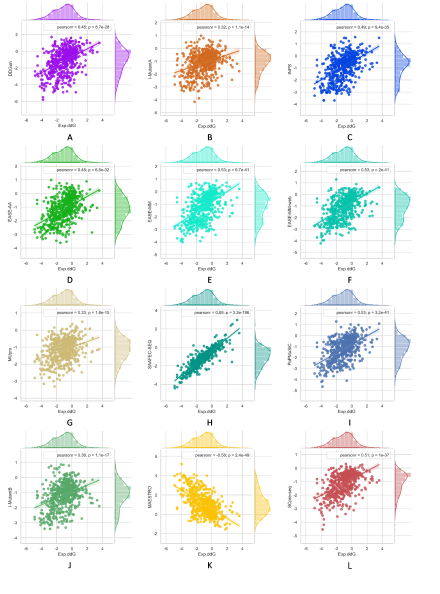
Figure 3. Multiple bivariate plots for 11 comparative methods and SCpre-seq with marginal histograms on dataset S543.
Casestudy
Predicting the impact of single-residue mutations on p53 thermodynamic stability
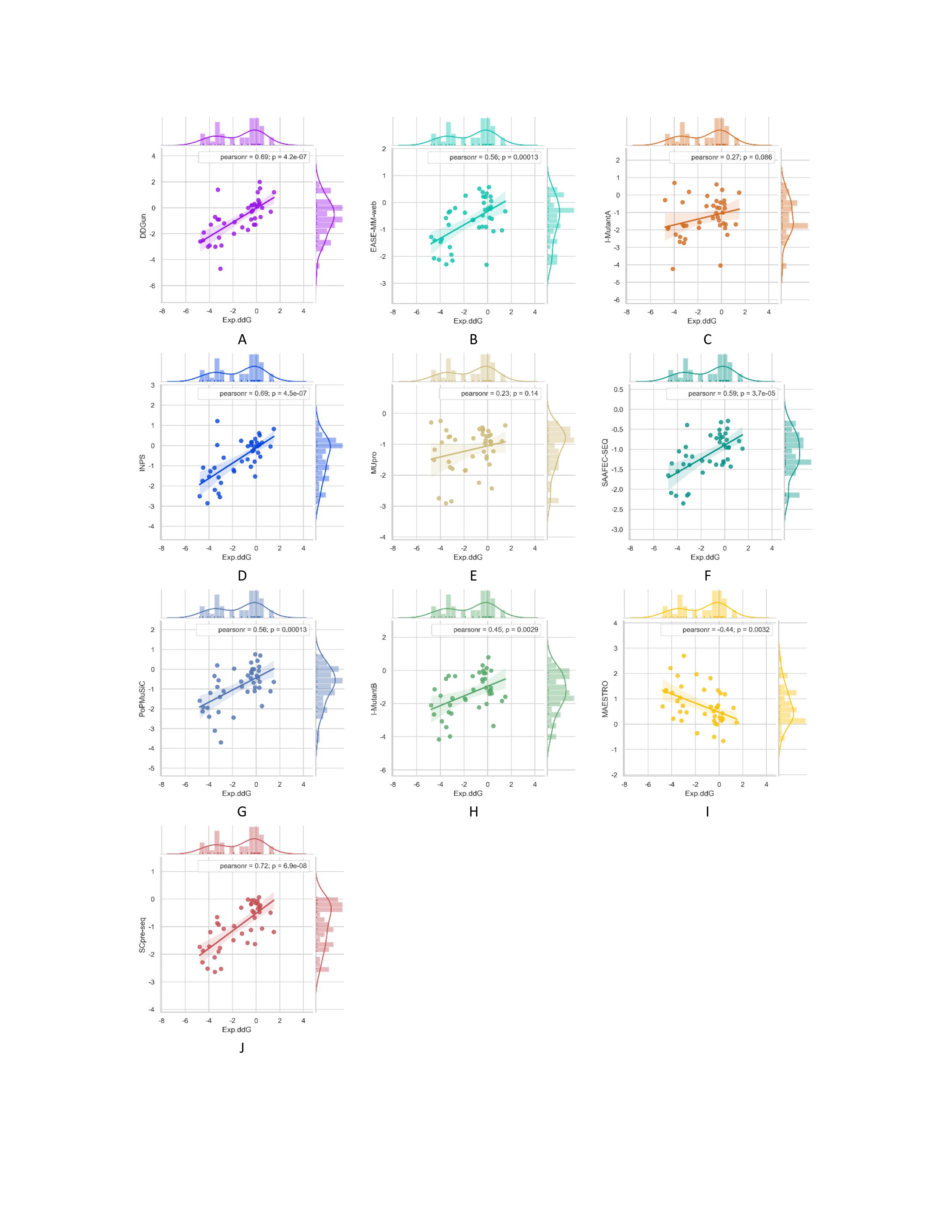
To further demonstrate the applicative power of SCpre-seq, we applied it to a disease-related protein p53 containing 42 mutations. The mutations include 31 stabilizing mutations and 11 destabilizing mutations. p53 case datasets can be downloaded from p53.
Multiple bivariate plots for nine comparative methods and SCpre-seq with marginal histograms. DDG predicted with the three structure-based methods (G, H, I) and six sequence-based methods (A, B, C, D, E, F) including SCpre-seq as function of experimentally measured stability changes (Exp.ddG) from the p53 dataset. The lines are the linear regression fits. Pearsonr represents Pearson correlation coefficient.
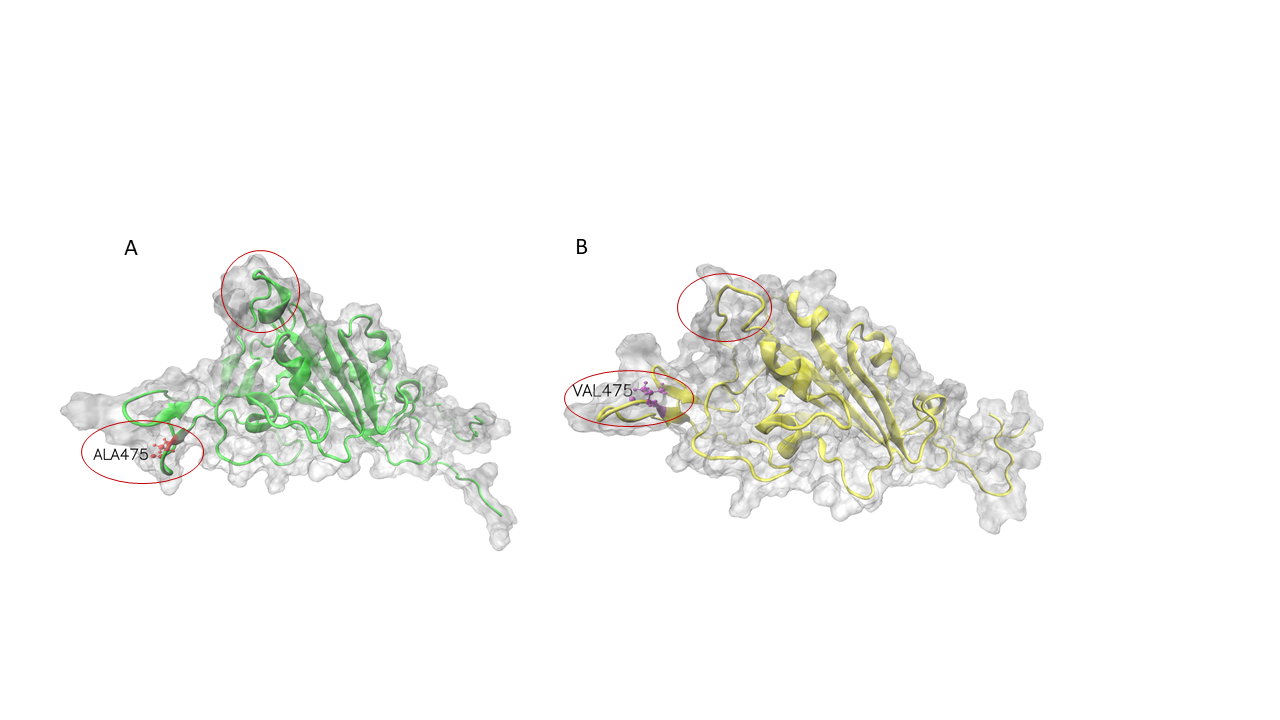
Mutation effects on for SARS-CoV-2 variants by stability changes perspective
The S proteins form homo-trimers on the virus surface and are necessary for the entrance of the virus into the host. Here, we only explored the effects of mutation on monomeric spike protein. The receptor-binding domain (RBD) is from 319 to 541 (UniProt ID: P0DTC2). Due to three PDB structures i.e. 6VXX (Closed state), 6VYB (Open state) and 6VSB (Prefusion) being discontinuous, we try to demonstrate wild-type and mutation RBD of spike protein 3D structures by AlphaFold2. The structures had visible changes after the A475V mutation and their structures align shown as follows.

Release
1.0
References
Please cite:
The codes are available at https://github.com/hurraygong/SCpre-seq
Contact
Jianting Gong: gongjt057@nenu.edu.cn
Juexin Wang: wangjue@missouri.edu
Dong Xu: xudong@missouri.edu